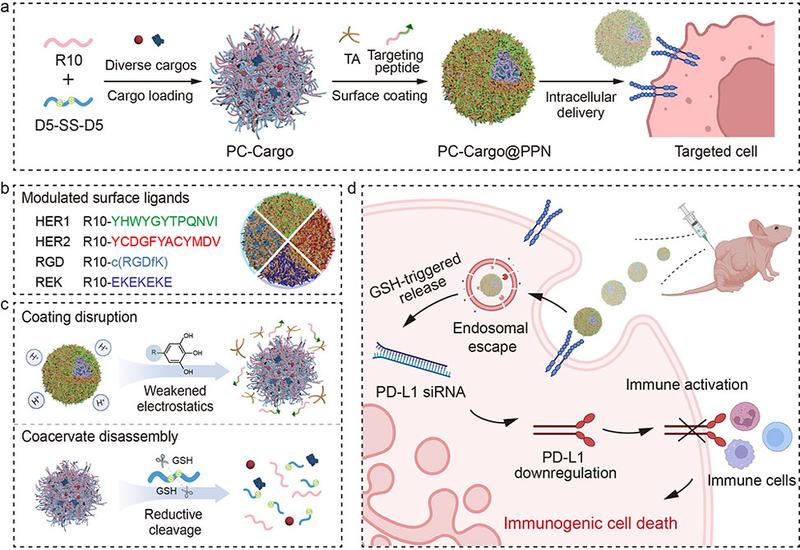
Delivering therapeutic biomolecules to the right cell at the right time remains one of the central unsolved problems in nanomedicine. Coacervates, concentrated liquid droplets formed by liquid–liquid phase separation, can encapsulate proteins, nucleic acids, and small molecules with high efficiency and without harsh processing conditions. Yet the same biophysical properties that make coacervates attractive cargo reservoirs also make them difficult to control: they coalesce in complex biological media, enter cells indiscriminately, and release their contents unpredictably. Engineering a surface coating that simultaneously confers stability, cell selectivity, and stimulus-triggered disassembly on a single coacervate platform has remained an open challenge.
Researchers in the Caruso Group at The University of Melbourne and the Jiang Group at Sichuan University, published in J. Am. Chem. Soc., constructed a modular system they call PC@PPNs, peptide coacervates, PCs, coated with polyphenol–peptide networks, PPNs. The core coacervate forms when a decaarginine peptide (R10) and a disulfide-bridged decaaspartate peptide (D5-SS-D5) are mixed under mild aqueous conditions. Tannic acid, TA, a natural polyphenol whose galloyl groups engage in versatile noncovalent interactions, then assembles with customized targeting peptides on the coacervate surface. By exchanging the peptide component of the PPN, the researchers programmed the surface with HER1-targeting, HER2-targeting, αvβ3 integrin-targeting RGD, or antifouling REK sequences, each producing a distinct surface charge without altering the spherical morphology or size of the particles.
The PPN coating transformed the physical behavior of the coacervates. Uncoated PCs coalesced within two hours in phosphate buffer, cell culture medium, or serum albumin solution, leaving no detectable particles. PC@PPNs survived the same conditions intact yet disassembled efficiently under reducing conditions: 81.1 ± 1.3% disassembly occurred at physiological glutathione, GSH, concentrations, and approximately 93% disassembly was observed at 5 mM GSH after two hours. Mass spectrometry confirmed that GSH cleaved the disulfide moiety in D5-SS-D5, fragmenting the peptide and collapsing the electrostatic interactions that hold the coacervate together. An independent acidic-pH trigger, arising from protonation of TA phenolic groups, provided a second disassembly pathway relevant to the endosomal environment. Loading efficiency reached up to 94.9 ± 0.1% for hydrophilic cargos including rhodamine B, fluorescein, IgG, and BSA. In cell studies, the TA component promoted endosomal escape through a proton-sponge mechanism, with minimal lysosomal colocalization confirmed by a Pearson's correlation coefficient of 0.45 ± 0.02. GSH depletion with L-buthionine-sulfoximine abolished cytosolic release, confirming that cargo liberation is reductively gated.
The versatility of the platform extended across a broad cargo range. PC@PPNs delivered saporin, a ribosome-inactivating protein that is membrane-impermeant in free form, to reduce CAL-27 cell viability to 49.2 ± 6.4% at 20 μg mL−1. The pro-apoptotic peptide KLA reduced viability to 32.6 ± 0.4% at 10 μg mL−1 and triggered mitochondrial depolarization as measured by JC-1 fluorescence shift. Functional enzymes including β-galactosidase (430 kDa), horseradish peroxidase, and Cre recombinase retained catalytic activity after cytosolic delivery. Cre-mediated loxP recombination switched reporter cells from red to green fluorescence, confirming nuclear translocation of the delivered protein. Plasmid DNA encoding mCherry or enhanced green fluorescent protein produced detectable expression after transfection via PC@PPNs. In a HER2-positive SKOV3 ovarian cancer model, HER2-targeted particles associated with 74.6% of tumor cells compared with 13.8% for antifouling-coated controls. Peritumoral delivery of doxorubicin-loaded PC@PPN-HER2 reduced tumor volume by 72.6% relative to saline and by 33.0% relative to the non-targeted formulation over two weeks, while free doxorubicin caused an 18.4% body weight loss that was absent in the targeted-coacervate groups. In a B16F10 melanoma model, RGD-targeted PC@PPNs carrying PD-L1 siRNA suppressed tumor growth by 78%, elevated mature dendritic cells in tumors to 26.6 ± 0.6%, and increased CD8+ T lymphocytes in lymph nodes approximately 4.1-fold relative to saline. RNA sequencing of treated tumors identified 807 differentially expressed genes, with downregulation of checkpoint molecules (Pdcd1, Cd274, Foxp3) and upregulation of effector immune genes (Prf1, Gzmb, Il2, Cd8a), consistent with a shift toward an immune-active tumor microenvironment.
The PC@PPN architecture separates coacervate core chemistry from surface identity, allowing targeting, stability, and release kinetics to be tuned independently by selecting different peptide ligands for the PPN layer. The authors suggest the platform is broadly applicable to cargo classes beyond those tested here and point to cancer immunotherapy as a near-term application area. The demonstration that a single coating strategy can confer receptor-selective uptake across HER1-, HER2-, and αvβ3 integrin-positive cancer models, while preserving the encapsulation efficiency and stimulus responsiveness that make coacervates attractive, provides a design framework that may inform next-generation biomolecular delivery systems.